
This is the first article in a series focusing on China Drug Master File (DMF) filing regulatory regulations and recent updates in the Chinese market.
Keywords: China DMF, API, Excipient, Packaging Material
Abstract
In the rapidly evolving landscape of pharmaceutical regulation in China, it is essential for companies seeking market entry to understand the intricacies of drug master file (DMF) filing. This article provides a detailed exploration of China’s regulatory framework, recent regulatory reforms, registration pathways and requirements associated with DMF filing in China. By shedding light on key considerations and recent developments, this article equips companies with the knowledge needed to navigate the intricate process of DMF filing in the dynamic pharmaceutical regulatory landscape of China.
China’s Regulatory Framework
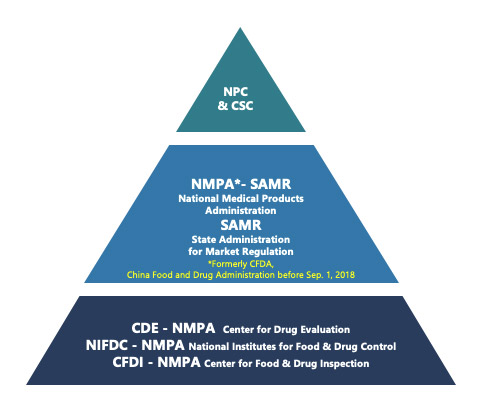
Regulatory framework in China (Figure 1)
The regulatory framework for pharmaceutical products in China is a multi-tiered structure, with various government departments playing essential roles. The pyramid (Figure 1) highlights the roles and tiers of these government departments.
Top Tier:
-
- China Congress (NPC) and the State Council (CSC): These bodies are responsible for drafting laws that form the foundation of the regulatory system.
Second Tier:
-
- National Medical Products Administration (NMPA): Formerly known as the China Food and Drug Administration (CFDA), the NMPA drafts regulations, oversees market approval, and conducts post-marketing surveillance.
Departments Under China NMPA:
-
- Center for Drug Evaluation (CDE): Responsible for drafting technical guidelines and reviewing application dossiers.
-
- National Institutes for Food and Drug Control (NIFDC): Handles quality testing.
-
- Center for Food and Drug Inspection (CFDI): Conducts on-site inspections.
Recent Regulatory Reforms
China’s drug regulatory framework has undergone significant reforms, particularly with the transition from the old Drug Master File (DMF) approval system to the new Drug Master Filing (DMF) system. Key aspects of these reforms include:
-
- Announcement No.146 (2017): This marked a significant policy reform, replacing the cumbersome approval process for active pharmaceutical ingredients (APIs), excipients, and packaging materials (AEP) with a more streamlined filing system.
-
- New Filing System: The streamlined China DMF filing system enables a binding review and approval process as part of drug registration, rather than requiring separate registration to obtain an import drug license. This policy realignment emphasizes the responsibility of marketing authorization holders (MAH) for ensuring the quality and safety of drug master files in China.
-
- International Alignment: The new China DMF policy aligns more closely with international systems, incorporating principles from ICH guidelines and drawing parallels with the EU ASMF and CEP systems, as well as the US DMF system. This alignment facilitates faster DMF registration and improved market access for international pharmaceutical companies entering the Chinese market.
Registration Pathways in China
Under the new China Drug Master File (DMF) filing system, there are three pathway options for Active Pharmaceutical Ingredient (API), excipient, and packaging material (AEP) approval:
-
- Binding Review and Approval Pathway: This is the most common pathway, involving a binding review and approval process through the Center for Drug Evaluation (CDE). The applicant prepares the DMF dossier and files an application to the CDE for a completeness review to obtain an inactive filing number. Subsequently, upon drug product registration (e.g., an abbreviated new drug application [ANDA] or a new drug application [NDA]), the CDE will refer to the inactive filing number, conduct a technical review of the DMF dossier along with the drug dossier, and then activate the DMF filing number.
-
- API Standalone Pathway: This pathway allows for standalone review and approval of APIs, similar to the EU Certificate of Suitability (CEP). The applicant obtains an inactive DMF filing number after a completeness review, and then the application proceeds to a technical review by the CDE for subsequent activation. This pathway is currently only applicable to generic APIs used in drug products that have been approved in China.
-
- Part of Drug Dossier Pathway: This pathway integrates the AEP dossier into the drug product dossier, emphasizing collaboration between suppliers and drug product applicants.
DMF Filing Workflow
The general steps and timeline for filing a Drug Master File (DMF) in China are as follows:
-
- Material Completeness Check: Assess the completeness of materials to ensure they are generally complete.
-
- Data-Gap Analysis: Conduct a detailed analysis on the technical content, providing guidance on supplementing missing data according to China CDE requirements.
-
- DMF Dossier Preparation: Compile the DMF dossier in Chinese and file it with the Center for Drug Evaluation (CDE).
-
- CDE Completeness Review: The CDE reviews the dossier for completeness and provides feedback within one to two weeks.
-
- Issuance of China Inactive DMF Filing Number: If the required materials are complete, the CDE issues the inactive filing number within one to two weeks on the official online platform.
-
- Technical Review: The CDE performs a technical review. The queuing time at the CDE may vary depending on the registration timeline of the associated drug product and its application category (e.g., new registration or change application). For APIs, the timeline is the same for new registrations and change applications.
-
- Activation of China DMF Filing Number: The CDE usually takes from 5 to 18 months to complete the technical review and activate the DMF filing number.
Timeline Overview
-
- Inactive DMF Number: It takes around two to five months to obtain a China DMF number (inactive status).
-
- Active DMF Number: It typically takes around 5 to 18 months for the technical review and activation of the DMF number. However, for the binding review and approval pathway, the timeline will depend on the drug product registration status, as the DMF is activated upon drug product approval. The timeline may be longer if there are delays in the quality testing for the registration of the API or drug product.
Understanding these steps and timelines is crucial for efficient DMF filing in China, ensuring compliance with regulatory requirements and timely market entry.
China DMF Checklist
The general China Drug Master File (DMF) material checklist for Active Pharmaceutical Ingredients (API), excipients, and packaging materials (AEP) is described in the table below:
|
API
 |
Excipient  |
Packaging  |
|
|---|---|---|---|
| • ICH M4 Module 1 Administration documents and drug information (including application form, etc.) | • Application form | • Application form | |
| • Applicant information | • Applicant information | ||
| • Product information | • Product information | ||
| 2.3.S & 3.2.S (ICH M4 CTD) | •General information | •Manufacturing information | •Manufacturing information |
| • Manufacture | • Characterisation | • Quality control | |
| • Characterisation | • Quality control | • Certificate of analysis (COA) | |
| •Control of drug substance | • Certificate of analysis (COA) | • Stability study | |
| •Reference standards or materials | • Stability study | • Compatibility study | |
| • Container closure System | • Pharmacology and toxicology study | • Safety study | |
| • …. | • …. | • …. | |
Preparing a Product Dossier for China DMF Filing: Before filing a DMF, the applicant needs to prepare a product dossier according to China regulatory requirements. Some of the requirements in the checklist are common across all categories of AEP, such as applicant information, certificates and a letter of authorisation.
Other items in the checklist are category specific. For example, API dossier is generally prepared as ICH M4 CTD format whereas excipient and packaging dossiers are compliant with Chinese regulations. For a detailed checklist according to your product, please send us an email: info@accestra.com.
Costs and timelines
Understanding the costs and timelines associated with the China Drug Master File (DMF) filing process is crucial for efficient planning and execution. The table below provides an overview of the administrative costs and average timelines for different product types:
| Product type | Administrative cost | Timeline (averages) |
|---|---|---|
| Active pharmaceutical ingredient (API) | $27-54k Or €25-50k | • Obtain DMF Inactive filing number: 2-5 months. |
| Excipient | Free | • Obtain DMF Active filing number: 5-18 months. |
| Packaging material | Free |
As China’s regulatory landscape continues to evolve, a thorough understanding of registration procedures and local requirements is essential for effectively navigating the drug master filing (DMF) process. By following the outlined steps and timelines, applicants can streamline the DMF filing process and obtain approval in the shortest time possible.
This article was prepared by April Wang from Accestra Consulting which provides China Regulatory Affairs Consulting and Drug Master Filing (DMF) for APIs, Excipients and Packaging Material.
Contact Us
For questions, please contact us by email: info@baipharm.com or visit www.dmfchina.com for more information.